 By Independent News Roundup
By Independent News Roundup

This post is very long as it contains my full written testimony for the Dutch court. The post is structured into:
As an introduction for those who are unaware of this case, this is a short video message from Peter Stassen, the only remaining attorney on the case. His colleague, Arno van Kessel, has been imprisoned on [fabricated] charges of terrorism and disbarred:
Expert witnesses: COVID shots ‘indistinguishable from bioweapons’
During the press conference, Stassen also noted his efforts to have the Dutch courts accept his expert witnesses’ in-person testimony. He said the witnesses intend to present evidence showing that the COVID-19 shots:
- Are “indistinguishable from bioweapons.”
- Offer “no health benefits whatsoever.”
- Are “neither safe nor effective.”
- Were released in the U.S. under emergency use authorization, “a legal status that removes the enforcement of the pharmaceutical law and consumer safeguards by the FDA,” or the U.S. Food and Drug Administration.
- Are “by design, intended to cause the damage described in the package insert and reports as ‘side effects’” — including, “sudden death, heart failure, cancer, and the most horrific diseases.”
- Are a “key component” of the “Great Reset,” “a military project in which NATO plays a significant role.”
This is my short video testimony, summarizing my written affidavit (provided in full minus 1G attachments, below):
Video summaries by Mike Yeadon, Katherine Watt, Catherine Austin Fitts and Joe Sansone, as well as other video interviews are available on this YouTube channel.
I usually try to avoid bringing unnecessary social media BS into my articles, but this information is very important and relavant, so it needs to be discussed. If you are a follower of Jikky Leaks (aka Mouse Piss) and associated “mouse army” (aka Moussad) on X, or a follower of Dr Ah Kahn Syed (same person(s) as Jikky on X), or OpenVAET (aka “canceledmouse” on X) and many other bullshit artists, named or anonymous trolls with mouse logos in profile - beware! They are a network of military-intel-private interest funded agents, and their job is to pick fights and “increase ambiguity” (intelligence term) by generating lots of seemingly relevant content, but the goal is to confuse and lead away from what is really pertinent in the investigation. For example, lead away from law/regulatory frameworks/military law into endless and unproductive “gold standard science” debates. This is often called “chaos agents” - chaos, or disorganization as opposed to cosmos, or structure and clarity. These ops also focus on smearing people who are on the right track about the covid op and, most importantly, are making some tangible progress. The agents named above pretend to be on the right side of this by posting lots of technically valid material, and criticizing vaccines. However, they insist that everyone goes after BigPharma only, and stay in the lane of the “gold standard science” debate. As my readers know, the real crime committed during covid has nothing to do with science. It had everything to do with the globally coordinated military attack on society, paid with military budgets/ contracts, ordering substances and protocols which are indistinguishable from weapons from the private sector accomplices in BigHealth/BigPharma.
Here is Jikky’s immediate lies in response to The Defender article which I linked above:
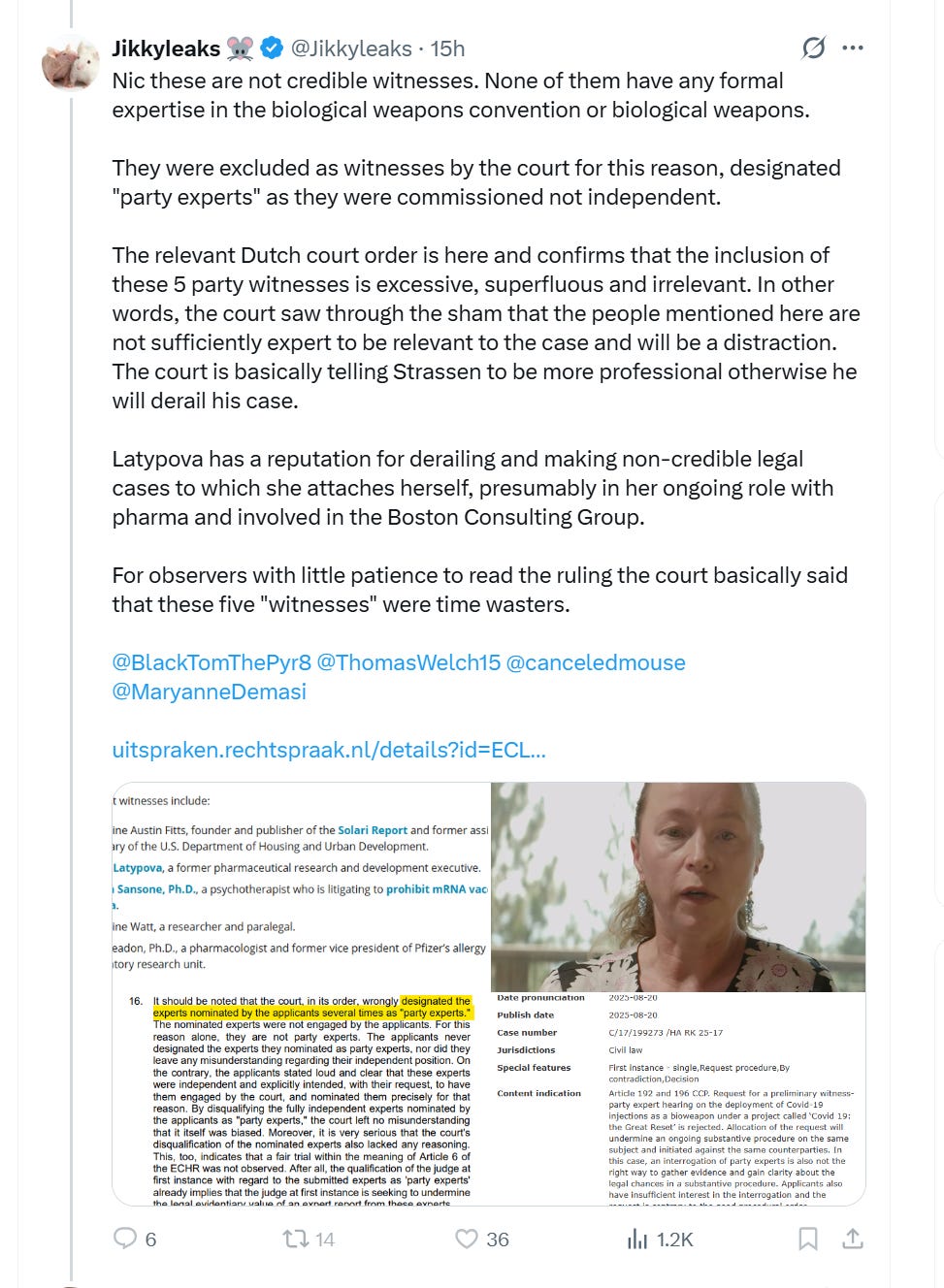
Note that what he writes about me is all lies:
Here is a collage of multiple Jikky Leaks, aka “Mouse Piss”, identities:

And here’s Jikky’s real identity - a psychotic, fake “doctor” whose specialty is bully and abuse women. He is not a real doctor however, he spends nearly 100% time online running trolling operations for whoever pays him to do that. Which is good news because he can’t physically hurt anyone any more:

Enough of this nonsense.
My written expert testimony, submitted to the court on September 15, 2025:
Scope and Expert Opinion:
Based on my review of primary regulatory documents, leaked Pfizer’s Chemistry‑Manufacturing‑Control (CMC) files, relevant legislation in the U.S. and EU, and other publicly available documentation, it is my expert opinion that the Covid‑19 mRNA injections were deployed under military ‘medical‑countermeasure’ rules that bypassed standard pharmaceutical safeguards, rendering them legally and functionally indistinguishable from a potential bio-chemical weapon.
Due Diligence and Art is a reader-supported publication. To receive new posts and support my work, consider becoming a free or paid subscriber.
1. The Dual‑Use Nature of mRNA/DNA Platforms
Established “dual‑use” designation – Since at least 1997, U.S. defense advisors (JASON group) and later the U.S. National Academies have listed gene‑therapy platforms, including lipid‑nanoparticle (LNP) mRNA systems utilized as vaccines, as technologies that can be weaponized (e.g., by delivering toxins, oncogenes, or immune‑suppressive micro‑RNAs).
Ease of weaponization – The same attributes that make mRNA attractive for therapy (cell entry, high expression) make it attractive for hostile use; even fragmented RNA (shRNA, miRNA) can dysregulate host gene expression without coding for proteins.
2. “Bait and Switch”: Consumers Worldwide Were Misled About the Legal Status of Covid-19 Products as Countermeasures – Medicines Used for Non-Medicinal Purposes, i.e. as Weapons.
Consumer safeguards normally in force were removed for Covid‑19 mRNA products:
Documentary Evidence of Non‑Compliance with Good Manufacturing Practices and Product Adulteration (Potential Weaponization):
3. Evidence that Covid Response Was Not a Public Health Response, but a Classified Military Operation Utilizing Medicines as Weapons
All Covid-19 countermeasures, including bio-chemical substances marketed as “safe and effective vaccines”, were ordered by the DoD as a “large scale manufacturing demonstration” via Other Transactions Authority contracts. According to Operation Warp Speed/HHS Assistant Secretary for Preparedness and Response (ASPR) reports, the US Department of Defense (DoD) ordered and oversaw the development, manufacture, and distribution of all countermeasures.
The Covid Dossier (Exhibit 1) is a compilation of the evidence from many countries and regions of the world demonstrating that:
Covid was not a public health event, although it was presented as such to the world’s population. It was a global operation, coordinated through public-private intelligence and military alliances and invoking laws designed for CBRN (chemical, biological, radiological, nuclear) weapons attacks.
The Dossier contains information regarding the military/intelligence coordination of the Covid biodefense response in the U.S., U.K., Australia, Canada, the Netherlands, Germany, Italy, and many more locations. For as many countries as possible, the Dossier lists the military/intelligence agencies in charge of their country’s Covid response; dates on which emergency declarations were made in each country; military/intelligence-related agencies and bodies in charge of censorship/propaganda; and top people with military/intelligence jobs who were known or reported to hold leadership positions in the response. The Dossier also lists connections to global governing bodies, including the EU and UN/WHO, through which the response was coordinated, and provides a listing of the military/intelligence/biodefense alliances that provided multinational frameworks for responding to a bioterror/bioweapons attack.
4. Covid-19 mRNA Injections are Indistinguishable from Bio-Chemical Weapons.
Prepared for counsel as foundational narrative; detailed citations, exhibits, and curriculum vitae available on request.
Biological and bio-chemical weapons are both naturally derived and man-made materials designed to induce disease through the introduction of toxins and microorganisms. The method through which a bio-chemical weapon is deployed depends on the agent itself, its preparation, its durability, and the route of administration. Attackers may disperse these agents through aerosols or food and water supplies[1]. In addition to externally dispersing such agents, introduction of bio-chemical agents via other means is described in literature dealing with bio-chemical weapons and terrorism. Specifically, use in consumer products, medicines, or even pet food and other products has been described as possible vectors of attack.[2]
In politics, diplomacy, and export control “dual use” refers to technology that can be used for both peaceful and military aims. mRNA/DNA technology, including embodiments as injectable drugs or vaccine products, has long been identified as a dual-use, potentially weaponizable technology[3],[4],[5]. The “dual use dilemma” was first noted with the discovery of the process for synthesizing and mass-producing ammonia, which revolutionized agriculture with modern fertilizers but also led to the creation of chemical weapons during World War I. The dilemma has long been recognized in chemistry and physics, leading to international conventions and treaties, including the Chemical Weapons Convention and the Treaty on the Non-Proliferation of Nuclear Weapons.
In 1997 the JASON group[6], an advisory committee to the US President on scientific matters pertaining to war technologies identified potential for genetically engineered pathogens in the following six groups:
Top of Form
Bottom of Form
To date, both awareness and control systems for detecting and preventing potential subversion and misuse remain woefully inadequate. Despite the obvious threat introduced by advanced synthetic biology products, if misused as weapons, little to no control measures exist today beyond a handful of guidance documents for scientists and research institutions. These self-reporting requirements are largely ignored, and as evidenced by the NIH grants to Wuhan Institute of Virology, research that is likely to raise objections is offshored.
In principle, any drug or injectable medical product can be weaponized, i.e., used as a poison instead of medicine. This is because the consumer or healthcare provider cannot directly assess the product and its ingredients and must rely on regulators enforcing pharmaceutical law/regulations to ensure compliance and safety as the product travels through the manufacturing supply chain and distribution. Pharmaceutical substances contain potentially dangerous chemicals, and typically, the difference between a drug and a poison/lethal weapon is the precise dosage, which the pharmaceutical regulations are designed to keep tightly controlled. As an example, consider opioids for legitimate use - pain management vs. their potential for lethal overdose. A mislabeled opioid product with an incorrect specification for dosage would be an example of a weaponized medicine.
According to the literature on bioweapons, weaponization of gene therapy/mRNA technology or another DNA/RNA platform technology can be accomplished in numerous ways. Synthetic mRNA is a large molecule (~3000+ base pairs). It is unstable and fragile, breaking into smaller segments during manufacture, storage, and transportation. It has been demonstrated that segments of RNA, such as short hairpin RNAs (shRNAs) or micro RNAs (miRNAs), can be exploited as weapons, and do not need to be precisely made, nor do they need to “code” for anything specific.
The following paragraph is found in Chapter 6 of the 2018 edition of the textbook “Biodefense in the Age of Synthetic Biology”[7]:
“Small RNAs are an example of functional genetic information that could be horizontally transferred. Small RNAs, although not a genome modification per se, are important because they may prove capable of modifying gene expression and bringing about phenotypic change. The large number of small interfering RNA (siRNA), short hairpin RNA (shRNA), micro RNA (miRNA) (Zhang et al., 2007; Huang et al., 2008), and other small-RNA library studies in a variety of species and cells from different species, including human, provides a potential roadmap of what sequences may lead to what disease states or to modulation of defenses against disease. {…}
One reason that RNA delivery is potentially a viable biological threat is that even a small initial skew in gene expression (such as the changes in gene expression normally caused by miRNAs) could greatly alter the probability of an initial cellular alteration. Even small amounts of a targeted RNA would not modify the genome per se, but might allow or encourage cells to begin the process of self-transformation to tumors, as evidenced by the fact that a large number of pro-oncogenic miRNAs have already been discovered (O’Bryan et al., 2017). In addition to RNAs produced by viruses, bacteria produce numerous small regulatory RNAs; introduction of these into the endogenous microbiome could lead to dysbiosis. Larger mRNAs can also be delivered via liposomes and nanoparticles or by RNA replication strategies being developed for vaccine production (see Chapter 8, Rapid Development of Self-Amplifying mRNA Vaccines); these methods could potentially be used to express deleterious cargo such as toxins or oncogenes, similar to threats related to DNA vectors.”
Clearly, both small and larger RNA sequences can be introduced into the human body for nefarious purposes, including the promotion of cancer, dysbiosis, immune system suppression, and organ damage. This can be accomplished with and without incorporation of the weaponized RNA code into the host genome and by RNA sequences that do not need to encode any protein.
Furthermore, the same textbook states that vaccine platforms are recognized a potential vehicle for weaponization:
“Larger mRNAs can also be delivered via liposomes and nanoparticles or by RNA replication strategies being developed for vaccine production [referring to self-amplifying mRNA vaccine platform] …these methods could potentially be used to express deleterious cargo such as toxins or oncogenes.”
Therefore, the mRNA/DNA products marketed as “vaccines” are, in principle, open to adulteration, whether intentional or due to a lack of proper manufacturing process controls and purity characterization. Technical capabilities that are required to reliably characterize and control these substances at the point of manufacture, in distribution, and administration, are nascent today and had not been routinely established by the manufacturers nor by the regulators in 2020-2021 when these substances were mass deployed worldwide.
In both the United States and the European Union, pharmaceutical regulations are designed to ensure that medicines are safe, effective, and of high quality before they reach the public. While there are many similarities between the U.S. and EU systems—especially in their reliance on scientific evidence—each has its own regulatory bodies and procedures for evaluating drugs and approving claims about safety and efficacy.
In the United States, the Food and Drug Administration (FDA), through its Center for Drug Evaluation and Research (CDER) and the Center for Biologics Evaluation and Research (CBER), is the primary authority responsible for approving pharmaceutical products. Before a new drug can be marketed, the sponsor must submit a New Drug Application (NDA) or Biologics License Application (BLA) that includes robust data from preclinical studies and clinical trials demonstrating the product’s safety and efficacy for its intended use. The clinical trial process requires investigational status of the product under Investigational Drug Exemption and follows a structured progression through Phases I–III, with formal oversight by an independent Investigational Review Board (IRB) ensuring protection of human subjects and execution of valid informed consent procedures. Only after this thorough process—culminating in a favorable risk-benefit assessment—can safety and efficacy claims be made. The FDA also reviews the manufacturer’s compliance with current Good Manufacturing and Clinical Practices (cGxP), assuring the product’s fidelity to the precise quantities of ingredients and labeling to ensure that all claims are substantiated and not misleading.
In the European Union, the regulatory landscape is governed by the European Medicines Agency (EMA) for centralized approvals, while national competent authorities (such as Germany’s BfArM, France’s ANSM or CBG/MEB in the Netherlands) handle decentralized or mutual recognition procedures. For most innovative drugs, especially those involving biotechnology or affecting multiple EU member states, the centralized procedure is mandatory. This involves submitting a Marketing Authorization Application (MAA), which must contain comprehensive data similar to what the FDA requires, including clinical and non-clinical findings. Safety and efficacy claims are scrutinized by the EMA’s Committee for Medicinal Products for Human Use (CHMP) before an opinion is issued and authorization is granted by the European Commission.
In both jurisdictions, post-marketing surveillance is essential. Companies are required to conduct pharmacovigilance activities and may be asked to perform additional studies (Phase IV trials) to monitor long-term safety and effectiveness. Any promotional material or advertising must strictly reflect the approved labeling and claims; unsubstantiated or misleading claims can lead to regulatory action. Additionally, there are stringent requirements for transparency, including public disclosure of clinical trial data.
While the U.S. and EU systems have distinct regulatory structures and processes, there is significant harmonization through international initiatives, such as the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH) and Mutual Recognition Agreements (discussed further in this testimony).
The pharmaceutical regulator in the Netherlands is the Medicines Evaluation Board (CBG/MEB). The CBG is responsible for assessing and monitoring the safety, efficacy, and quality of human and veterinary medicines in the Netherlands. It evaluates marketing authorization applications (MAAs) for new drugs and ensures that product information and claims are accurate, evidence-based, and compliant with regulatory requirements. The agency also participates in the European regulatory network, working closely with the European Medicines Agency (EMA) and other national authorities in the EU.
CBG’s responsibilities include:
The CBG operates under the Dutch Ministry of Health, Welfare and Sport and collaborates with the Health and Youth Care Inspectorate (IGJ), which enforces compliance with pharmaceutical laws and inspects manufacturers and healthcare providers.
The actions of the US FDA are directly related to those of the EMA due to the Mutual Recognition Agreements that national food and drug regulators signed in the years leading up to 2020. These agreements are contracts that allow one regulator to accept, without further review, what another regulator in a different country or region has presumably examined and approved. This legal framework was created to promote harmonization and efficiency. However, during the orchestrated global COVID event, this structure was exploited by the perpetrators for malicious purposes, enabling a false facade of regulation by the FDA and allowing other regulatory agencies to simply accept FDA’s statements without conducting mandatory independent reviews of the data.
Here is one MRA as an example, between the FDA and EMA.[8] This US-EU MRA was entered into force on 1 November 2017 for human medicines and 30 May 2023 for veterinary products. It became fully operational for human medicines as of 11 July 2019:
“qualified persons in the EU Member States do not need to batch test human medicines covered by the MRA, provided that they have verified that these controls have been carried out in the United States for products manufactured in and imported from the United States.”
Most, if not all, other national food and drug regulatory systems now rely on the FDA regulations and their compliance/cGMP monitoring, without conducting any of their own reviews, regulation, batch testing, or other cGMP procedures.
While the Covid-19 mRNA injections were broadly marketed as “safe and effective vaccines,” all Covid products and protocols entered the global markets without following the usual pharmaceutical manufacturing and distribution compliance standards for regulated drugs, due to their legal status as “countermeasures under a public health emergency.” In other words, they are considered non-medicinal. This status is managed by a completely different, militarized governance framework and a separate set of laws, originally designed to respond to time- and location-specific attacks involving Chemical, Biological, Radiological, and Nuclear (CBRN) weapons. The defendants were aware of this. The public has not been informed of the true legal status of these injections or their permitted use.
US FDA provides the following definition of medical countermeasures:
“Medical countermeasures (MCMs) are FDA-regulated products such as biologics, drugs, and devices that may be used in public health emergencies to diagnose, prevent, or treat diseases or conditions caused by CBRN threats or emerging infectious diseases.”[9]
While the statement on the FDA website claims that MCMs are “FDA-regulated”, this statement is misleading because it omits the fact that MCMs are not regulated like typical pharmaceutical products, as expected by consumers and healthcare providers.
Under U.S. federal law and EU law, for medicinal uses of medicinal products, FDA/EMA must formally approve any new investigational drug before a manufacturer can introduce it into interstate commerce.
This process requires the manufacturer to submit an Investigational New Drug application and obtain approval from the FDA/EMA for its use in regulated clinical research (trials). This regulated process is therefore called an “investigational” regulatory pathway. It mandates that a manufacturer conduct regulated clinical research trials, obtain Institutional Review Board’s (IRB) approval for clinical trial protocols, ensure independent safety monitoring, and secure informed consent from clinical trial volunteers. Additionally, the manufacture of drugs and biologics under investigational status must strictly comply with current Good Manufacturing Practices (cGMP) and broader cGxP regulations.
CMs can include both previously FDA/EMA approved medical products (e.g. opioids, ventilators, antibiotics, masks, swabs, etc), and new unapproved products (e.g. mRNA/DNA injections). However, the key distinction between MCMs and normally regulated medicines is the statutory non-investigational categorization of MCMs. Specifically, in US law, use of Emergency Use Authorized (EUA) covered countermeasures under a declared Public Health Emergency cannot constitute a clinical investigation (21 USC 360bbb-3(k)), therefore countermeasures cannot be tested for safety or efficacy in accordance with US law (21 CFR 312 and 21 CFR 601), nor can compliance with current Good Manufacturing Practices (cGMP) or Good Distribution Practices (GxP in general) be enforced by the FDA.
This re-categorization of previously approved and unapproved medical products deploys MCMs for “unapproved uses”. Together with the non-investigational status, this re-categorization precludes the use of MCMs as medicines, and, by removing consumer safeguards, leaves MCMs entirely open to weaponization.
The laws that enable the removal of all consumer safeguards and manufacturers’ liability include the PREP Act (2005)[10], Sec 564 of the Food, Drug and Cosmetics Act (FDCA)[11] in the US and a set of EU provisions for “pandemic preparedness” and “medical countermeasures” (discussed in Section 2.5). The PREP Act and corresponding EU provisions for countermeasures waive enforcement of pharmaceutical law for products declared countermeasures under an existing emergency declaration and provide liability immunity to covered persons (except narrowly defined willful misconduct). The PREP Act law is subject to legal controversy, and a proposal for its repeal is currently pending in the US Congress[12].
Due to the declared non-investigational status of MCMs, while the manufacturers may choose and the FDA/EMA may ask to undertake some of the activities typically expected from an investigational clinical trial and manufacturing validation process, none of the typical pharmaceutical regulatory standards are applicable in an enforceable way. In general, any activities claimed as regulated investigational tests and processes for medical countermeasures should be deemed deceptive practices designed to manufacture a veneer of consumer protection where none exists, nor intended to exist.
FDA has the discretion to issue an EUA if, in the sole opinion of the HHS secretary, the product “may be effective” in treating the relevant disease or condition[13]. No other criteria for approval apply in an enforceable way. There is no strict legal requirement to conduct clinical trials prior to authorization and no lawful human subject-protected clinical trials are possible due to statutory “non-investigational” status of countermeasures.
FDA will approve EUA countermeasures on incomplete/non-existent information based on an opinion of the HHS Secretary that “known and potential benefit of the product” may “outweigh the known and potential risks”[14] and considers it unlikely that “comprehensive effectiveness data” will be available before an EUA grant. In contrast, for an investigational drug (under normal regulatory approval process) the FDA “shall” deny approval if the applicant “do[es] not show that such drug is safe.”[15]
There is no strict requirement for an Investigational New Drug exemption (IND), nor institutional review board (IRB) approval of a clinical trial protocol and informed consent forms. Therefore, the EUA status of a medical countermeasure precludes collection of the regulated clinical trial data and thus precludes reliable, valid scientific knowledge of risks and benefits associated with the EUA Countermeasure while it remains non-investigational.
Furthermore, there are no required standards for quality-control in manufacturing; no inspections of manufacturing procedures; no lot-release testing and no prohibition on wide variability among lots; no prohibition on adulteration; and no required compliance with Current Good Manufacturing Practices. EUA products, even though unregulated and non-standardized, “shall not be deemed adulterated or misbranded.”[16]
The EUA pathway for medical countermeasures is used only when the United States Secretary of Health and Human Services or Minister of Health in EU Member State(s) declares an emergency[17]. In the United States, the PREP Act emergency declarations for covid have been extended 12 times to last until end of 2029[18], preventing enforceable regulations and extending the liability shield for deaths and injuries caused by the countermeasures.
In the Netherlands, the government formally classified COVID‑19 as a Category A infectious disease under its national Public Health Act (Wet publieke gezondheid) on January 27, 2020. This classification — announced by Health Minister Bruno Bruins —activated a range of emergency measures. This step is effectively the national declaration of a public health emergency, empowering authorities under the Public Health Act. These powers enabled nationwide responses—such as lockdowns—without declaring a formal emergency status. The Netherlands never invoked a formal national “public health emergency” declaration; therefore, there’s no emergency status to formally end.
In summary, in both US and EU, legal status of a product, service, procedure or action designated as “countermeasure” is equivalent to that of a potential weapon. Medicinal products or potential medicinal products (unapproved drugs), when designated as “countermeasures” under real or fabricated emergency, are legally permitted to be used for non-medicinal purposes. The non-medicinal purposes include use as a weapon or an illegal human experiment. Misrepresentations of safety, efficacy, or contents of EUA products are allowed by the applicable law. The defendants knew or should have known this, yet they concealed this from the public and propagated monstrous lies and coercion to achieve the maximum scope of deployment of these pretend medicines, which are in fact bio-chemical weapons.
While the relevant laws between US and EU are not identical, there are several EU provisions corresponding with the provisions in US in relation to “medical countermeasures”.
In the European Union (EU), medical countermeasures (MCMs) are regulated primarily through a framework of EU pharmaceutical, medical device, and public health laws, especially as they relate to preparedness for serious cross-border health threats. While the EU doesn’t use the term “medical countermeasure” as explicitly as the U.S. FDA does, the legal infrastructure supports the development, authorization, stockpiling, and deployment of such products in public health emergencies.
Key EU Laws and Regulations Governing Medical Countermeasures:
1. Regulation (EU) 2022/2371 “On serious cross-border threats to health”
Adopted: 2022
Establishes:
2. Regulation (EU) 2022/123 “On a reinforced role for the European Medicines Agency (EMA)”
Adopted: January 2022
Purpose: Expands EMA’s role in monitoring, coordinating, and facilitating the development and availability of medicines and medical devices during public health emergencies.
3. Joint Procurement Agreement (JPA) under Article 5 of Decision 1082/2013/EU
4. In 2016 the EU enacted a Regulation (EU) 2016/369, https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32016R0369&from=EN , which provides for emergency support within the European Union. Emergency support “may be awarded through specific measures appropriate to the economic situation in the event of an ongoing or potential natural or man-made disaster.” (Art. 1 (1)) Based on a decision of the Council according to Art. 2 of Regulation 2016/369/EU “to activate the emergency support” under this Regulation, after proposal submitted by the EU Commission, the stage has been set for financing “specific measures” against threat emerging from “human made or natural disasters”.
On 14 April 2020 this Regulation was amended by Regulation (EU) 2020/521[19] with retroactive effect as of 1 February 2020, in order to extend the application of this Regulation to emergency support during Covid-19 crisis.
In its Art. 1 the “emergency support under Council Regulation (EU) 2016/369” is activated.
This Regulation provides for an Annex of Regulation 2016/369/EU in which it enumerates the eligible measures which may be funded in case of emergency situations. It says that the enumeration is not exhaustive, and it reads:
“The following actions may be financed in case of pandemics with large‐scale effect:
(a) temporary reinforcement of the medical workforce, exchange of medical professionals, hosting foreign patients or other type of mutual support;
(b) deployment of temporary healthcare facilities and temporary extension of existing healthcare facilities to relieve pressure on existing structures and increase overall healthcare capacity;
(c) activities to support the administration of large‐scale application of medical tests and prepare the necessary scientific testing strategies and protocols;
(d) setting up temporary quarantine facilities and other appropriate measures at the Union borders;
(e) development, production or purchase and distribution of medical products;
(f) increases and conversions of production capacities for medical products as referred to in point (e) to address supply shortages;
(g) maintenance of the stock of medical products as referred to in point (e) and their disposal;
(h) actions to support the necessary steps to obtain approval for the use of the medical products as referred to in point
(e) if required;
(i) actions to develop appropriate methods to track the development of the pandemic and the results of measures
implemented to address it;
(j) organisation of ad‐hoc clinical trials of potential therapies or diagnostics according to trial standards agreed at Union level;
(k) scientific validation of medical products, including potential new testing methods.
The above list is not exhaustive.’
Thus, (h) enables any actions asserted necessary to achieve the approval for the use of medical products. It enables avoiding any provisions applicable to medicinal products /drugs and medical devices.
Lit (j) shows that for “ad-hoc” clinical trials GCP (Good Clinical/ Good Manufacturing Practice laws) can be waived and replaced with rather vaguely defined “trial standards agreed at Union level”, meaning the rules agreed upon by unelected bureaucrats, rather the pharmaceutical regulations spelled out in the law that governs consumer safety in medical products.
Extensive evidence of public deception by pharmaceutical companies acting in concert with the regulators became available in late November 2020, when approximately 1000 pages of Chemistry Manufacturing Controls (CMC) documentation and a set of internal email exchanges were leaked from EMA[20]. The full set of leaked pages and emails is included in the Attachment to this affidavit. These documents demonstrated that public health officials in the European Union committed fraud on all citizens, giving the impression that they evaluated and approved the covid injections according to existing standards for medicinal products, at least based on conditional marketing authorizations (CMA). The documents revealed that behind the scenes regulators were concerned solely with the timing of the launch, even before any data was reviewed, and waiving or making up greatly reduced quality standards to help Pfizer “meet” those standards, based on Pfizer’s say-so and no scientific basis whatsoever. This deceptive “bait and switch” scheme was coordinated globally among the regulatory agencies – the FDA, EMA, MHRA, TGA, Health Canada and many others.
In the EU, the same net effect – absence of any enforceable compliance with pharmaceutical cGxP regulations and other relevant pharmaceutical law - has been accomplished by forcing all Member States to sign EU-orchestrated predatory purchasing contracts with pharmaceutical manufacturers that waived all relevant pharmaceutical regulations and obligated national governments to indemnify the manufacturers in case of successful liability suits against pharma companies in the Member States. This indemnification included waiver of sovereign immunity (see p.32 of Pfizer Advanced Purchase Agreement with EU[21]). These contracts effectively prevented the Member States from exercising sovereign legislative powers with respect to pharmaceutical liability for these products in their countries. These contracts have been allegedly negotiated by Ursula von den Leyen by text messages. There is an ongoing investigation by the European Public Prosecutor’s Office (EPPO) into the acquisition of COVID-19 vaccines in the European Union[22]. The defendants who acted as corresponding health authorities in the Netherlands knew or should have known that these contract clauses were predatory, violate the respective state’s Constitution, and thus are unacceptable for a sovereign government, and would result in mass injuries among the citizens these public health officials had sworn the oath to protect.
One of the critical mechanisms of deception was tying all 27 EU Member States into one (blind) deal by promising that the vaccines in Europe will go through the Conditional Marketing Approval (CMA) regulatory pathway as opposed to the Emergency Authorization, i.e. the issue of Art 5(2) vs CMA. This is discussed in several emails included in the EMA documentation leak. For example:

The Art 5(2) is an Emergency Authorization mechanism available individually to the Member States. This authorization is issued for one year only, it is issued by each state and each state can revoke it independently. The CMA is a pan-EU mechanism in which the individual states have no say. The CMA in EU, however, is a non-emergency investigational pathway which, prior to 2020, had only been used for oncology drugs as a “right to try” or “compassionate use” for terminally ill patients. It is similar to the Expanded Access Use[23] (EAU, not to be confused with the EUA) in the US. Both CMA and EAU pathways however are investigational, meaning legally designated as medicinal use and purpose of the product. However, as discussed above, the “countermeasures” are non-investigational substances designated for non-medicinal purposes, thus coloring them as CMA or “BLA” is fraud and deception associated with their use as weapons.
As it is spelled out in law, the CMA sounds like a much stronger regulation and compliance mechanism vs Art5(2):

The important difference between Art 5(2) and CMA is that it puts liability onto the manufacturer, and that’s what was promised to be enforced via the use of the pan-EU CMA authorization:

However, in a classic bait-and-switch fraudulent inducement scheme the public health authorities in the EU and the Netherlands never meant to hold the pharmas to any of those promised CMA standards. The EU supply contracts with pharmaceutical companies waived all the relevant consumer safety enforcement regulations and laws in their countries. In effect, this removed any meaning from the CMA standards for consumer safety and manufacturer liability, because no enforcement = no law!
For example, see the EU supply agreement with Pfizer[24] wrt Indemnification:
In summary, the net effect of the PREP Act (i.e. removal of all relevant pharmaceutical regulations and manufacturers’ liability) was accomplished in EU via centralizing supply agreements under the pan-EU fraudulent scheme designed to pass military countermeasures onto the consumers under the color of CMA-authorized and later “fully approved” vaccines.
Approximately 1000 pages of Pfizer’s manufacturing documentation were leaked from the European Medicines Agency (EMA) at the end of 2020 showing absence of the Good Manufacturing Practice (cGMP) compliance less than 2 weeks before the product was deployed onto billions of consumers worldwide. The leak of the documents was authenticated by the British Medical Journal. The EMA did not deny the authenticity of the documents. In addition to the manufacturing documentation, the EMA files also contain 14 screenshots of emails dating from mid to late November 2020. The email exchanges are between the EMA staff and senior executives at the agency and their correspondence with the FDA and MHRA (the UK regulator). These emails demonstrate that the EMA reviewers were under massive political pressure to invent new ways of approving the inherently non-approvable dangerous products. It is evident from the emails, comments and objections raised by the EMA review staff that they were not aware of the legal status of the Covid-19 injections as countermeasures, nor that the regulatory review of the data was not material to their deployment.
It is also evident that the EMA leadership were concerned primarily with coordinating the launch dates, and the “authorization” of these shots for all EU Member States was a forgone conclusion. The pressure to overlook all regulatory deficiencies was emanating from the very top of the US, UK and EU governments.
There were severe and unresolvable - given the purposefully unrealistic timeline - issues with the quality of the product the EMA staff were pressured to ok. The manufacturing process was woefully out of compliance. The EMA reviewers raised more than 100 objections to approval, including several Major Objections. Specifically, Major Objections included
1) lack of cGMP compliance;
2) lack of mRNA integrity and large amounts of mRNA fragments (some of which can be characterized as miRNA, siRNA and shRNA - all potential weaponization components as described above);
3) many significant gaps in manufacturing documentation making it impossible to determine if the product could be made as described.
Emails leaked from EMA also demonstrate that the EU regulators were only concerned with the dates of product launch and were not going to review the necessary data prior to launch. The process was highly political and not scientific. The EMA regulators took verbal assurances from the FDA officials instead of reviewing the data. As stated above, the EMA staff reviewers objected to the data, but the high-ranking officials ignored and over-ruled their concerns.
Lack of cGMP compliance means that no assurances can be made that the products contain specific ingredients in specific amounts in the units dispensed to the patients. Therefore, in addition to the product being dangerous, no informed consent is possible.
One of the key elements guaranteeing the quality of the drug substance is a product specification based on the current state of product development as well as science and technology. For this, the framework is set by International Conference on Harmonization (ICH) guideline Q6A “Test procedures and acceptance criteria for new drug substances and new drug products: chemical substances”[25]. The specification defines a list of tests along with references to analytical procedures and appropriate acceptance criteria for the quality assessment of the respective product regarding identity, assay/quantity, purity/impurities, and potency/ biological activity.
All analytical procedures must be fully validated, which means that either standard, previously validated methods are used, or if proprietary/novel – then the manufacturer must invent and fully validate the assays that are used for novel techniques. This requires, among other things, definitive tests with positive and negative controls as well as full and traceable documentation “audit trail” for every test. Commercial secrecy is not an excuse for failure to comply with these requirements. Even if the manufacturer does not wish to publicly disclose their analytical procedures, they still must submit full transparent documentation to the FDA who holds it on file (and this fact is formally communicated to purchasers of the product for their own GxP compliance).
In Pfizer’s CMC documentation leaked at the end of 2020, the reviewers from EMA noted that there was no description of the “non-compendial”, i.e. Pfizer’s own developed proprietary methods for analytical procedures. This made it impossible to evaluate the scientific accuracy of the proprietary methods used by Pfizer to control the quality of produced injections.
Numerous product and process control parameters were proprietary, not well defined, some were not yet invented, and none were scientifically validated. For example, the test used for the mRNA identity testing was chosen RT-PCR (real-time PCR). However, the mRNA is a highly unstable and fragile substance, which, while manipulated and chemically “optimized” for stability, still has shown a great degree of fragility. As a result, the integrity of the mRNA has been problematic, especially with the scale up of manufacturing.
One of the Major Objections (MO) from the EMA reviewers stated in the leaked Pfizer CMC documentation was lack of mRNA integrity, i.e. low % of RNA in the vial conforming to the specification and a very high % of broken RNA pieces:

The large amount of non-conforming RNA was deemed impurity by the EMA reviewers. It is, indeed, a major problem - the entire claimed efficacy of the product allegedly depended on the “correct code” to produce the “Wuhan spike protein” using the cellular machinery of the injected individuals. Here we have evidence of the regulators objecting to approval due to the fact that many other things besides the “Wuhan spike protein” can happen in the cells bombarded with broke RNA pieces, which is a known weaponization method for mRNA vaccines as discussed above.
Instead of stopping the approval and demanding that the mRNA integrity is resolved, the regulators simply arbitrarily moved the acceptance criterium for the %mRNA integrity from the previous standard of 70%+ to just greater than 50%.
That means that a large portion of drug substance is allowed to contain “junk” RNA material, fragments, and uncharacterized pieces, some of them large enough to code for unknown and possibly aberrant proteins, and most of them falling into the category of micro RNAs (miRNAs). These short sequences, while non-coding, are known to interfere with cellular processes and are implicated in cancer pathways[26].
Ultimately, the regulatory review itself and the objections raised by the EMA reviewers did not matter - the product was going to be pushed on the market regardless of the regulatory objections due to the military CBRN response that was invoked globally, in secret from the public.
Neither the FDA nor EMA conducted the manufacturing facilities inspections for Pfizer and its suppliers in 2020, citing covid emergency. When the manufacturing facility inspections resumed in 2022, Pfizer’s major European contractor, Rentschler, was found in violation of cGMP (form 483 issued). This means the supply chain for Pfizer in Europe was not in compliance between 2020 and 2022. No enforcement action was taken by the regulators, as per EUA law there is no enforcement possible.
It is also worth noting that in 2019, evidently in preparation for the Covid-19 operation, then US FDA Commissioner Scott Gottlieb changed the federal regulations governing inspection of licensed facilities manufacturing all biological products including ‘vaccines’, from at least every two years to unspecified times; eliminated enforcement provisions if a licensed facility failed an inspection; and eliminated all inspection duties for FDA inspectors. Prior to the rule change, 21 CFR 600.21, Time of inspection, read:
“The inspection of an establishment for which a biologics license application is pending need not be made until the establishment is in operation and is manufacturing the complete product for which a biologics license is desired.
In case the license is denied following inspection for the original license, no reinspection need be made until assurance has been received that the faulty conditions which were the basis of the denial have been corrected. An inspection of each licensed establishment and its additional location(s) shall be made at least once every 2 years. Inspections may be made with or without notice, and shall be made during regular business hours unless otherwise directed.”
Effective May 2, 2019, the last three sentences of 21 CFR 600.21 were removed.
There is currently no legal requirement for an initial FDA inspection; no minimum interval for subsequent FDA inspections, and there are no legal consequences for compliance failures, such as establishment or product license denial or revocation.
The legal mechanisms through which FDA regulation of biological product manufacturing disappeared, included a Direct Final Rule and a Proposed Rule, simultaneously issued by Federal Register notice on Feb. 26, 2018, and an April 2, 2019 Final Rule, issued by then-FDA Commissioner Scott Gottlieb.
Regarding the technology platform itself (mRNA in lipid nanoparticle [LNP], or DNA in adenoviral vector) - both are known as “transfection” technologies. The purpose of the product design is to deliver various bio-chemical cargo inside the cellular membranes, and often into the cell’s nucleus where DNA resides. Transfection methods using a wide variety of RNA and DNA technologies are a well-established scientific reality. For example, as published in this literature review paper[27]:
“A systematic literature search based on PRISMA guidelines (Moher et al., 2009) was established to identify relevant published studies or protocols that fit into this review’s scope (Fig. 1). Databases that were employed for the literature search included Scopus, Google Scholar, and PubMed. The keywords being used during the search included “transfection”, “co-transfection”, “chemicals”, “reagents”, “DNA”, “siRNA”, “shRNA”, “miRNA”, “plasmid”, “oligonucleotides”, “efficiency”, “safety”, “cytotoxicity”, “controls” and other related key terms. An initial search returned about 5,000 articles, published protocols, or handbooks from various databases that reported the descriptions or comparisons between different transfection methods, types of transfected nucleic acids, transfection control, transfection efficiency assessment methods and transfection reagents.”
The design of the genetic therapy products is identical to that of the covid “vaccines”: the LNPs will deliver the cargo attached to them into the cells, i.e. transfect the cells. The LNP platform or adenovirus platform are just 2 different types of “cargo trucks”, the LNP being particularly effective in hacking the cells. The fact that it is a transfection technology is documented very widely in science literature and in regulatory documents. Both Moderna and BioNTech characterized their mRNA technologies as experimental, a “therapy” and in early development stages in SEC filings immediately preceding the pandemic. Moderna’s 2019 Form 10-K specifically noted: “mRNA medicines are a novel and unproven approach... No mRNA immunotherapy has been approved, and none may ever be approved.”
No mRNA pharmaceutical products were approved by any regulatory authorities in the world before 2020. This was due to numerous failures to meet pharmaceutical safety and compliance standards. All products in development repeatedly ran into safety issues and could not advance even into the first phase of the human testing[28],[29].
Covid operation was not a public health event, although it was presented as such to the world’s population. It was a global operation, coordinated through public-private intelligence and military alliances and invoking laws designed for CBRN (chemical, biological, radiological, nuclear) weapons attacks.
In the US, the National Security Council (and not Department of Health and Human Services) is in charge of Covid policy in the US. In other countries of the world, covid campaign was coordinated via identical mechanisms, with military and intelligence apparatus leading the response policy (as in a war), and public health agencies presenting a false front of a public health emergency to the public. See Exhibit 1 for detailed evidence of the global military campaign, including the evidence for the Netherlands and other EU countries.
In the US, March 13, 2020: “PanCAP Adapted U.S. Government COVID-19 Response Plan” (PanCAP-A) states that United States policy in response to SARS-CoV-2 is set not by the public health agencies designated in pandemic preparedness protocols (Pandemic and All Hazards Preparedness Act,[30] PPD-44,[31] BIA), but rather by the National Security Council, or NSC. NSC does not have regular attendees from public health agencies and its focus is national security and foreign policy matters.”
Below is the organization chart from the PanCAP-A document, p.9:

According to the Operation Warp Speed/ASPR reports, Operation Warp Speed was declared as a “collaborative” effort of the DOD and HHS to produce “safe and effective” Covid-19 vaccines and therapeutics. However, according to the organizational chart, the DOD was formally the Chief Operating Officer, while HHS had the Chief Science Advisor position.[32]

VRBPAC-10.22.20-Meeting-Presentation-COVID19-Vaccine-Development-Portfolio.pdf
Notably, the next seniormost layer of the organization is controlled by the US Government and includes all supervisory roles for manufacturing, clinical trial design and implementation, planning operations and analysis, distribution, public affairs, contracting, legal and other functions. The pharmaceutical companies are the third level down in this organization.
A report by STAT News in 2020 pointed out that roughly 60 military officials, including four generals, were involved in the leadership of Operation Warp Speed, many of them without any previous healthcare experience. Out of roughly 90 leadership positions on the organizational chart, only 29 were not employed by the DoD.[33]
The unclassified October 2020 documents from Operation Warp Speed presentations at the FDA’s Vaccines and Related Biological Products Advisory Committee reveal control of the US Government over nearly all product design and implementation aspects of the development for Covid countermeasures[34].
Hundreds of Covid countermeasures contracts became available via ”freedom of information” (FOIA) requests in partially redacted form.[35] Review of these contracts indicates a high degree of control by the US Government (DoD/BARDA) and specifies the scope of deliverables as “demonstrations” and “prototypes” only. Demonstration is a fake, performative activity. Medicinal products administered to people cannot be characterized as “demonstrations and prototypes” however, weapons can be ordered as prototypes. The contracts also include the removal of all liability for the manufacturers and any contractors along the supply and distribution chain under the 2005 PREP Act and related federal legislation.
While the DoD/BARDA countermeasure contracts refer to safety and efficacy requirements for vaccines and mention current Good Manufacturing Practices (cGMP) compliance, these items are explicitly carved out as not being paid (or ordered) by the US Government.
The contracts were structured under Other Transactions Authority (OTA) - a method of contracting that was utilized by the DoD and BARDA for all Covid-related countermeasures ordered from the private industry. The OTA method of contracting allows federal agencies to order otherwise-regulated products bypassing any such regulations, as well as financial accountability mechanisms that cover standard government contracting, and other laws that regulate disclosure and Intellectual Property (IP) derived from publicly funded research.[36]
“Other” is a catchall category that is not a contract, not a research grant, not a procurement, etc.: not any normally regulated/accountable government contracting.
DoD used OTA to order vaguely defined “prototypes” and “demonstrations” that are not subject to regulatory scrutiny.
BARDA’s own report, lists Covid-19 products as “demonstrations” or at best “large scale manufacturing”:

The DoD/BARDA contracts for “countermeasures” are managed by Advanced Technology International (ATI).[37] ATI mostly manages R&D consortia for the Department of Defense for things like weapons manufacturing, metal casting and forging, ship production and technology aimed at “countering Weapons of Mass Destruction (WMDs).” Two of these consortia related to biomedical projects.
The Medical Technology Enterprise Consortium (MTEC), operating on behalf of the U.S. Army Medical Research and Development Command, includes technology for gene-editing, nanotechnology, “telehealth solutions,” artificial limbs and brain implants. MTEC is currently developing a wearable device to diagnose Covid-19 before symptoms appear.
The Medical CBRN Defense Consortium (MCDC)[38] currently includes ~300+ large and small businesses and academic entities that “support the Department of Defense’s (DoD) medical pharmaceutical and diagnostic requirements to counter Chemical, Biological, Radiological and Nuclear (CBRN) threat agents” and enable “prototype technologies for therapeutic medical countermeasures targeting viral, bacterial and biological toxin targets of interest to the DoD,” including the development of vaccines.
Through the mechanism of Other Transactions Authority, MCDC contracted with hundreds of companies to deliver Covid-related “countermeasures.” Pfizer doses were ordered on July 20, 2020, through Base Agreement between Advanced Technologies Inc (ATI, a DOD vendor management company) and Pfizer, Inc., identified as MCDC Base Agreement No. 2020-532:
· July 21, 2020, MCDC Technical Direction Letter or Statement of Work (SOW) for “COVID-19 Pandemic - Large Scale Vaccine Manufacturing Demonstration” between Pfizer and DOD/Advanced Technologies Inc.[39]
The contracts specified that the product will be shipped to the DoD as sole purchaser. The delivered product is not serialized – i.e., unit doses are not barcoded and thus not traceable under normal pharmaceutical distribution rules which exist to flag any safety or quality issue in the supply chain. The product thus is open to both falsification/mislabeling and adulteration. The product was shipped to DoD and handled through a “black box” DoD distribution system, ostensibly due to the cold chain storage requirements, which were later removed, but the distribution practice via military contractors and military contracts did not change.
The product was specified as “US Government property”[40] until it is injected into a person. All persons performing any tasks along manufacturing, supply chain, distribution and administration of the shots are “covered persons” under PREP Act liability shields, as long as they follow US Government orders. Regardless of place of employment, they are deemed to be US Government employees for purposes of this work.
Importantly, the DOD contracts describe Covid countermeasures as intended for “civil and military application.”
Based on my review of primary regulatory documents, leaked Pfizer’s Chemistry‑Manufacturing‑Control (CMC) files, relevant legislation in the U.S. and EU, and other publicly available documentation, it is my expert opinion that the Covid‑19 mRNA injections were deployed under military ‘medical‑countermeasure’ rules that bypassed standard pharmaceutical safeguards, rendering them legally and functionally indistinguishable from potential bio-chemical weapons.
Covid-19 injections were deceptively presented to the public under color of medicines, falsely advertised as “safe and effective vaccines”.
Individuals who prescribed, purchased and/or administered the Covid-19 (mRNA) injections participated in war crimes and/or genocide (democide).
[1] https://en.m.wikipedia.org/wiki/List_of_U.S._biological_weapons_topics
[2] https://www.ncbi.nlm.nih.gov/books/NBK535870/
[3] https://sites.dartmouth.edu/dujs/2013/03/10/genetically-engineered-bioweapons-a-new-breed-of-weapons-for-modern-warfare/
[4] https://www.ncbi.nlm.nih.gov/books/NBK535887/
[5] https://irp.fas.org/threat/cbw/nextgen.pdf
[6] https://isgp-studies.com/jason-group-national-security-science
[7] https://www.ncbi.nlm.nih.gov/books/NBK535870/
[8] https://www.ema.europa.eu/en/human-regulatory-overview/research-and-development/compliance-research-and-development/good-manufacturing-practice/mutual-recognition-agreements-mra
[9] https://www.fda.gov/emergency-preparedness-and-response/about-mcmi/medical-countermeasures
[10] https://aspr.hhs.gov/legal/PREPact/pages/default.aspx
[11] https://www.fda.gov/regulatory-information/federal-food-drug-and-cosmetic-act-fdc-act/fdc-act-chapter-v-drugs-and-devices
[12] https://massie.house.gov/news/documentsingle.aspx?DocumentID=395737
[13] 21 U.S.C. § 360bbb-3(c)(2)(A)
[14] 21 U.S.C. § 360bbb3(c)(2)(B)
[15] 21 U.S.C. § 355(d)(2); See also 42 U.S.C. § 262(a)(2)(RB) (biologic approved only if it actually “is . . . safe”).
[16] 21 USC 360bbb-3a(c).
[17] 21 U.S.C. § 360bbb-3(a)(1), (b).
[18] https://public-inspection.federalregister.gov/2024-29108.pdf
[19] https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32020R0521&from=EN
[20] https://www.bmj.com/content/372/bmj.n627
[21] https://dn721909.ca.archive.org/0/items/contract_03/Contract%203_text.pdf
[22] https://www.eppo.europa.eu/en/media/news/ongoing-eppo-investigation-acquisition-covid-19-vaccines-eu
[23] https://www.fda.gov/news-events/public-health-focus/expanded-access
[24] https://dn721909.ca.archive.org/0/items/contract_03/Contract%203_text.pdf
[25] https://www.fda.gov/regulatory-information/search-fda-guidance-documents/q6a-specifications-test-procedures-and-acceptance-criteria-new-drug-substances-and-new-drug-products
[26] https://www.nature.com/articles/sigtrans20154
[27] https://pmc.ncbi.nlm.nih.gov/articles/PMC8067914/
[28] https://www.statnews.com/2017/01/10/moderna-trouble-mrna/
[29] https://thewashingtonstandard.com/moderna-a-company-in-need-of-a-hail-mary/
[30] https://www.govinfo.gov/content/pkg/PLAW-109publ417/pdf/PLAW-109publ417.pdf
[31] https://www.in.gov/dhs/files/FEMA-Fact-Sheet-COVID-Response-3.4.20.pdf
[32] See “VRBPAC-10.22.20-Meeting-Presentation-COVID19-Vaccine-Development-Portfolio.pdf” in Attachment
[33] https://www.statnews.com/pharmalot/2020/09/28/pharmalittle-operation-warp-speed-is-more-army-than-science-jjs-covid-19-vaccine-moves-forward/
[34] See “VRBPAC-10.22.20-Meeting-Presentation-COVID19-Vaccine-Development-Portfolio.pdf” in Attachment
[35] https://www.keionline.org/covid-contracts
[36] https://www.keionline.org/bn-2020-3
https://www.ati.org/
[38] https://www.medcbrn.org/current-members/
[39] https://www.keionline.org/misc-docs/DOD-ATI-Pfizer-Technical-Direction-Letter-OTA-W15QKN-16-9-1002-21July2020.pdf
[40] https://www.cdc.gov/vaccines/covid-19/vaccination-provider-support.html#6-23-22
[41] The Dutch government disclosed a secret obligation to comply with NATO Resilience goals (news article): https://deanderekrant.nl/kabinet-erkent-navo-is-de-baas/
